PyPWDFT: pure-python plane-wave density functional theory


PyPWDFT is a pure-Python package for performing plane-wave DFT calculations. PyPWDFT is designed with the aim of teaching students the inner workings of a PW-DFT code. The source code is kept relatively small and contains extensive commenting. Using PyPWDFT is fairly simple, though its application is due to its scope relatively limited.
Features and scope
Unit cells are limited to cubes.
There are no pseudo-potentials, nor any \(\vec{k}\)-point sampling outside of the \(\Gamma\)-point.
Lowest Kohn-Sham states are found using the Arnoldi iterative procedure.
Slater exchange functional
Vosko-Wilk-Nusair correlation functional (VWN5)
Dualism: the same basis set is used to describe both the molecular orbitals as well as the electron density.
A
pypwdft.SystemBuilderclass is used for quick generation of example structures.The self-consistent field procedure allows for verbose (detailed output)
Variables and matrices relevant to the computation are accessible to the user such that they can follow the procedure.
Example
The code below will perform a plane-wave density functional theory calculation for the methane molecule inside a 10x10x10 a.u. unit cell.
# import the required libraries for the test
from pypwdft import PyPWDFT, PeriodicSystem, MoleculeBuilder
import numpy as np
def main():
# create cubic periodic system with lattice size of 10 Bohr
npts = 16 # number of grid points
sz = 10
# construct CH4 molecule system via SystemBuilder
s = SystemBuilder().from_name('CH4', sz=sz, npts=npts)
# construct calculator object
calculator = PyPWDFT(s)
# perform self-consistent field procedure and store results in res object
res = calculator.scf(tol=1e-1, verbose=True)
if __name__ == '__main__':
main()
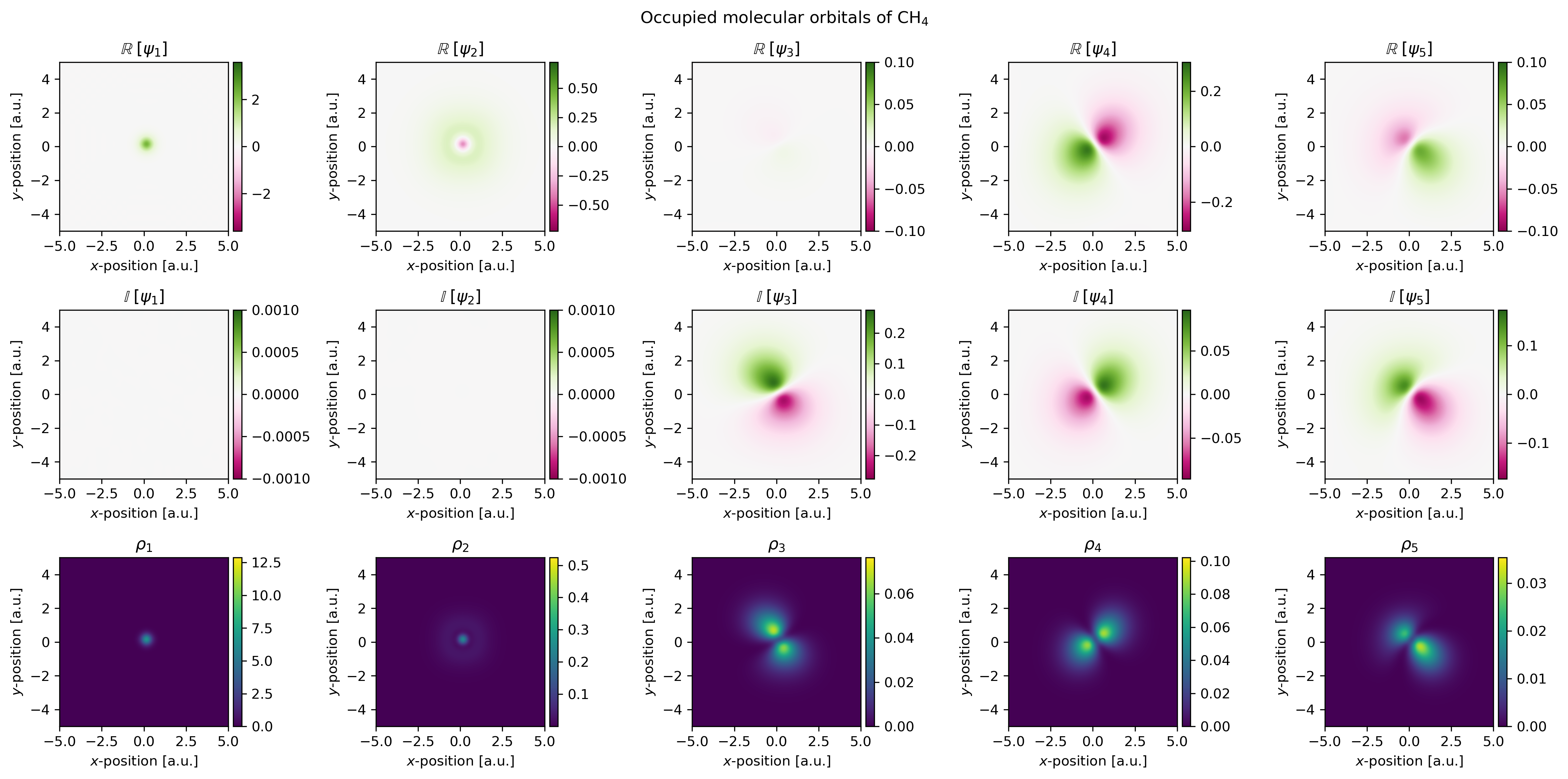
The set of molecular orbitals obtained via the above calculation are shown below. The top row corresponds to the real part of the molecular orbitals, the middle row to the imaginary part and finally the bottom row corresponds to the electron density associated with each molecular orbital.
PyPWDFT has been developed at the Eindhoven University of Technology, Netherlands. PyPWDFT and its development are hosted on Github. Bugs and feature requests are ideally submitted via the github issue tracker.